信息中心



北京时间2024年3月25日,复旦大学化学系刘智攀教授团队在Nature Catalysis期刊上发表了一篇题为“Square-pyramidal Subsurface Oxygen [Ag4OAg] Drives Selective Ethene Epoxidation on Silver”的研究成果(https://doi.org/10.1038/s41929-024-01135-2)。
该成果报道了基于机器学习原子模拟探寻银催化乙烯环氧化活性位点的最新研究成果,发现环氧乙烷产物选择性与反应条件下Ag(100)原位生长的含有五配位次表层氧的新表面相有关的理论和实验证据。
论文通讯作者是刘智攀教授,陈东晓、陈林为论文的共同第一作者。项目感谢自然科学基金委科学中心项目(12188101)、科学探索奖和复旦大学化学系实验室建设支持。
银催化乙烯环氧化反应是一个典型的选择性异相催化反应,具有较为成熟工艺路线和庞大的经济市场,但反应涉及的基础物理化学问题仍尚未解决。这其中,困扰研究者多年的问题便是在工业反应对应的高温高压条件下银催化剂表面结构究竟如何,以及是怎样的化学环境和反应机理能够使得乙烯和氧气最终选择性化合产生环氧乙烷。从实验的角度讲,由于现有技术无法在高压高温条件下获取清晰的表征图像,而只能获取模糊的振动谱学信息,因此了解催化剂在高压高温条件下的原位结构和反应信息极其困难。而从第一性原理计算和模拟的角度讲,探寻催化剂表面结构不仅需要处理各种不同的构型构象,还需要探索庞大的组分空间和各种可能的表面周期性,这使得传统方法,如基于密度泛函计算的巨正则系综的路径模拟或组分遍历方法,难以应对如此规模的复杂体系。
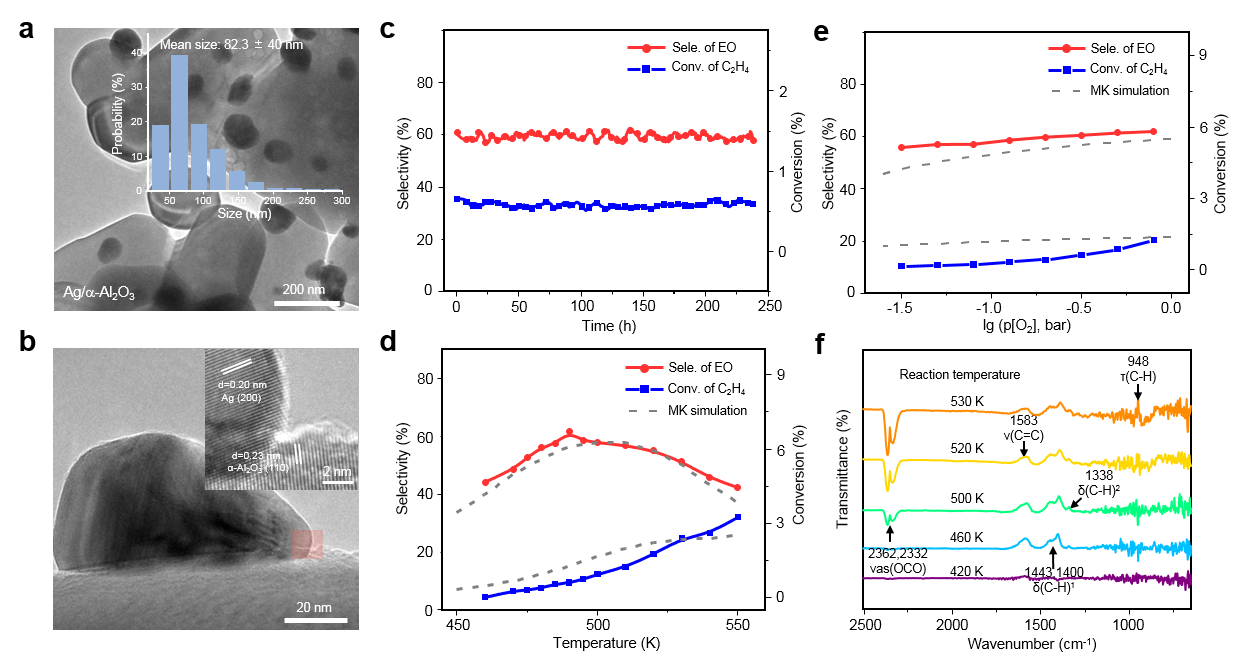
在这项工作中,作者使用了多项基于课题组(zpliu.fudan.edu.cn)前期开发的系列算法和程序,主要包括LASP大规模机器学习原子模拟 (www.lasphub.com),机器学习最优表面结构自动搜索(ASOP)算法和反应路径自动采样算法(MMLPS),对Ag(100)、Ag(111)、Ag(110)三个表面进行全局探索。得益于全局机器学习势函数在势能面计算上的稳定和速度优势,作者通过搜索三百多万个不同构型的AgxOy(C2H4)z表面结构,确定了三个银表面在反应条件(500 K,1 bar乙烯和1 bar氧气)附近的最优表面结构,发现具有四个共存相(图1)。其中,生长在Ag(100)的O5相具有表层氧、五配位的次表层氧[Ag4OAg],还包含特殊电子性质的强吸附乙烯(图2),而其他三个共存相只有表层氧。随后作者使用反应路径自动采样算法探索了乙烯在这些表面上的反应机理,发现只有O5相有利于环氧乙烷的形成(图3),而其它相的存在则是优先燃烧反应到CO2,从而抑制环氧乙烷选择性。在理论结构和机理的指导下,作者随后制备了Ag/a-Al2O3催化剂并进行原位实验(图4),发现490 K、1 bar乙烯和1 bar氧气的条件下,催化剂具有大于200小时的稳定的高环氧乙烷选择性(59.4 %)。原位的红外光谱表征发现了位于1583 cm-1的具有红外活性的碳碳双键伸缩振动峰,与理论对O5相上强吸附的乙烯进行红外模拟的结果(图2)类似,印证了理论对O5相活性位点的探测。另外,实验得到的动力学数据也与完全基于理论数据进行的微观动力学模拟结果一致,印证了理论对反应机理的预测。这一工作为揭示工业条件下复杂、动态的催化剂结构演化和催化活性关系提供了参考方案,促进了研究者在原子级别上对催化剂进行设计与调控的过程。
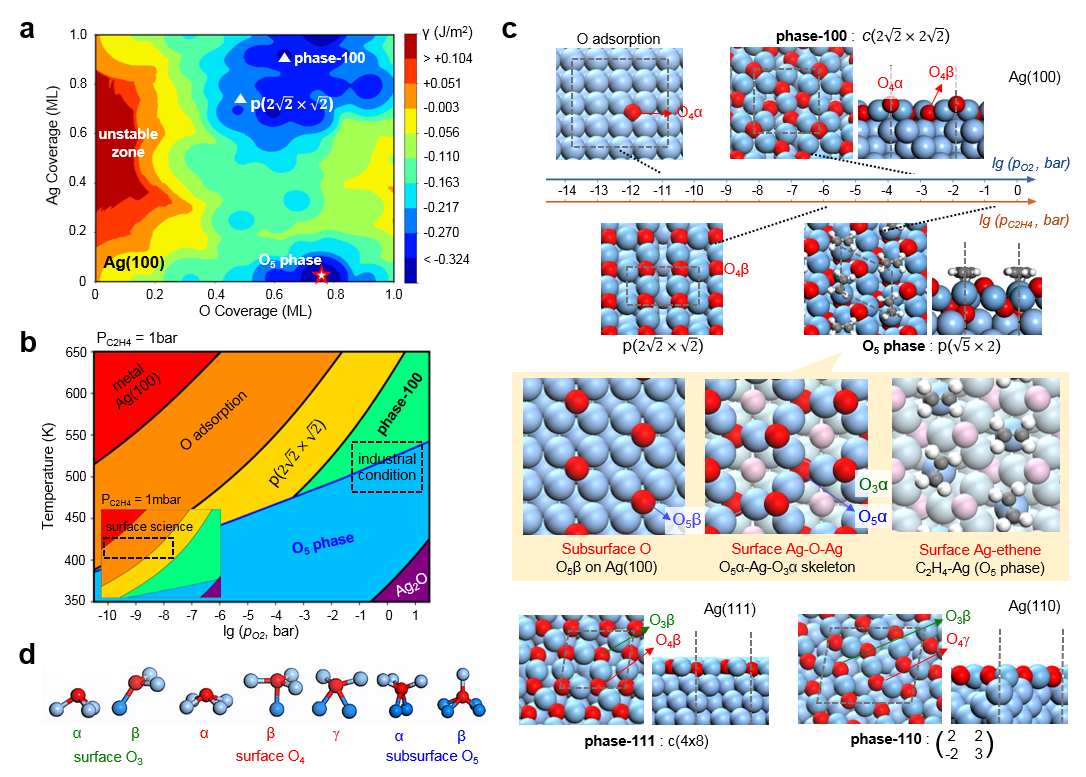
图1:ASOP算法获取银在反应条件下的最优表面结构。
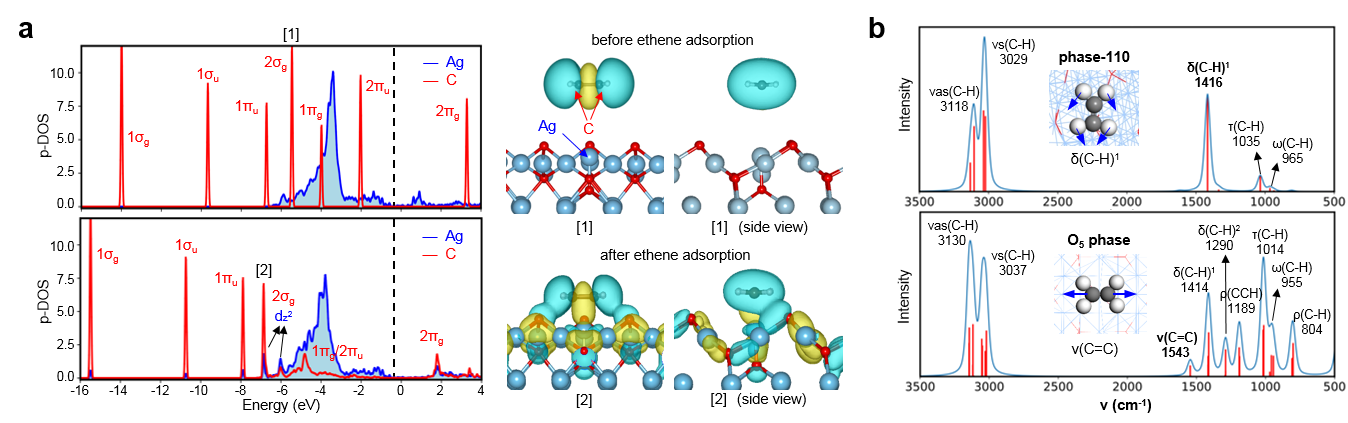
图2:O5相上强吸附乙烯的电子结构分析。
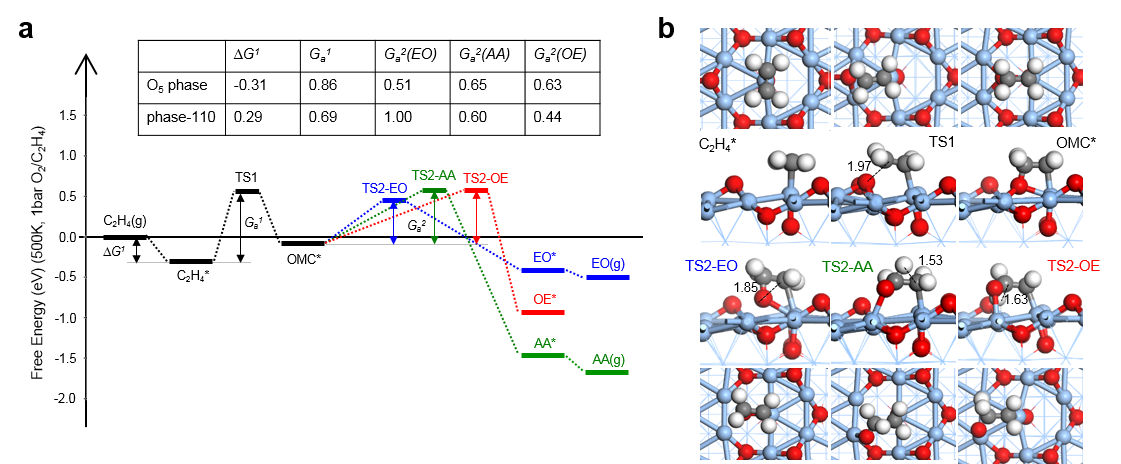
图3:反应条件下各个共存相的乙烯环氧化反应机理计算。