信息中心



如何通过理论化学计算预测并指导复杂工业催化实验,是极具挑战的科学问题,相应的研究始终伴随着催化科学的发展。随着大规模神经网络计算模拟的兴起,特别是刘智攀课题组开发的基于LASP软件的全局神经网络方法的发展和成熟,理论计算和化学实验的鸿沟得到了进一步的打破。近日,刘智攀课题组针对乙炔半氢化(C2H2 + H2 → C2H4)这个基础石油工业的重要反应,证明了全局神经网络方法计算和传统催化实验结合的新研究模式具有重要价值和意义,在前期复杂催化活性位结构搜索(Nature Catal. 2019, 2, 671)和自动反应预测(J. Am. Chem. Soc., 2019, 141, 20525)工作的基础上取得重要进展。
PdAg合金是乙炔半氢化最常用的工业低温催化剂,相比于纯Pd催化剂在选择性上有很大提升,但Ag元素的催化作用一直存在着争议,同时工业上低温的催化活性和选择性仍然不能令人满意(如,活性在50%时选择性大于90%)。刘智攀课题组结合机器学习模拟和催化实验阐明了PdAg合金催化剂在反应条件下的表面状态,并通过筛选发现了负载在金红石TiO2上的Pd1Ag3纳米催化剂,具有很高的催化性能(70 oC,乙炔转化率>96%,乙烯选择性>85%,稳定保持超过100小时,见图e-f)。
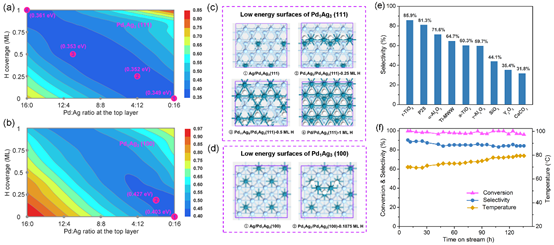
该工作从理论出发,通过神经网络全局结构搜索确定体相Pd-Ag-H相图和稳定的PdAg合金结构,并系统研究了稳定合金在反应条件下的表面相图(见图a-b)和稳定表面构型(见图c-d)。研究发现PdAg只有两个结构不同的关键体相Pd1Ag1 (R-3m)、Pd1Ag3 (Pm-3m),它们在反应条件下(25 oC,p(H2) = 0.05 atm)具有不同的表面结构,当Pd/Ag原子比 < 1/3时,(100)面才会被Ag全部覆盖;随着H2分压上升表面Pd会富集,在Pd1Ag3比例下富集主要出现在(111)面。进一步的反应路径搜索和微观动力学模拟揭示,乙炔加氢的活性受(111)面上的PdAg局部结构图案控制,选择性则主要由(100)面暴露的Pd的数量决定,在Pd1Ag3比例下能获得理论上的最高选择性(> 90%)。随后的催化实验证实了Pd1Ag3纳米颗粒的高催化性能,并通过观察体系在相同高转化率条件下的反应温度变化,确定了Pd在(111)面富集的趋势。最后,通过载体筛选发现金红石TiO2衬底能够细化Pd1Ag3纳米颗粒,降低Pd暴露在(100)面,从而获得更高的加氢选择性。
该工作由复旦大学化学系博士后李晓天(理论部分)和博士生陈林(实验部分)在刘智攀老师的指导下共同完成,期间得到了商城老师的帮助。该工作得到了国家重点研究计划纳米专项(2018YFA0208600)和国家自然科学资金(22033003, 21533001 and 91745201)的资助,和复旦大学化学系在实验室建设的支持。研究成果以“In Situ Surface Structures of PdAg Catalyst and Their Influence on Acetylene Semihydrogenation Revealed by Machine Learning and Experiment”为题发表在化学旗舰期刊Journal of the American Chemical Society上。
全文链接: https://pubs.acs.org/doi/10.1021/jacs.1c02471


