信息中心



机器学习势目前已经成为化学、物理、生物和材料科学等领域开展计算模拟的重要工具,也是人工智能技术在科学领域中应用的关键方向之一。目前,主流的构建方案基于原子能量分解或多体展开理论,旨在实现对复杂高维体系相对高效准确的描述。然而,基于原子展开得到的原子能量缺乏实际化学意义,而多体展开方法通常面临高阶展开项致计算复杂度指数增长的挑战。
近日,复旦大学化学系郁琦课题组提出了一种全新的机器学习势构建框架-MB-PIPNet,成功实现了势函数模型在匹配第一性原理精度的同时,能与传统分子力场方法计算效率高度融合。相关研究成果以“Extending atomic decomposition and many-body representation with a chemistry-motivated approach to machine learning potentials”为题,在Nature Computational Science上发表。
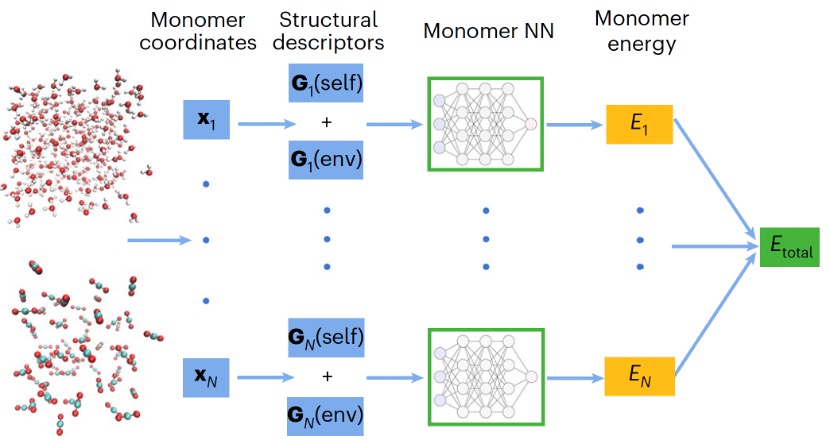
该研究不再采用传统的原子能量展开方案,选择了全新的路径,将复杂体系的总势能分解成有效分子单体能量(effective monomeric energy)之和。类比于电子结构理论中原子轨道能量与分子轨道能量的关系,有效分子单体能量具有更好的化学意义,能够直接描述分子自身结构变化及其对外界环境的响应程度。同时,课题组提出的MB-PIPNet算法目前无需复杂的神经网络架构。通过结合高效的交换不变多项式理论(PIP),仅需生成分子单体自身结构的描述符及其与不同单体之间的两体作用描述符,即可高效地包含整个体系中的多体相互作用。这一方案使MB-PIPNet算法在保证模型精度的同时,具备显著的计算优势:计算量与分子单体数目成正比,且无需引入复杂的三体等高阶描述符。
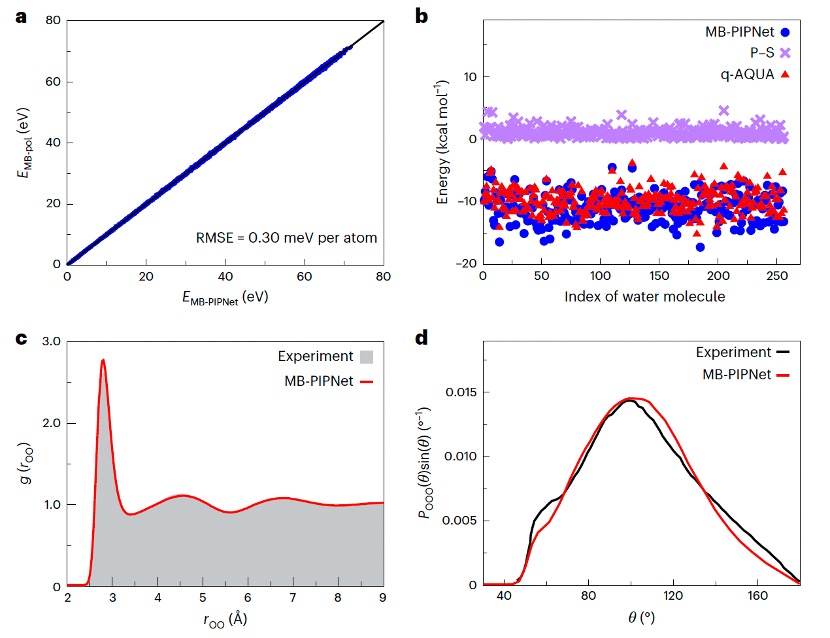
目前,MB-PIPNet框架已在不同气相和凝聚相复杂体系中进行了系统测试,为开发兼具传统力场速度和第一性原理精度的机器学习势提供了全新的思路。这一进展有望在有限计算资源下,对气相团簇、分子液体、生物大分子等体系实现长时间、多尺度的经典与量子动力学模拟。课题组正在探索前沿的机器学习网络架构,进一步改进发展MB-PIPNet相关算法。
复旦大学郁琦青年研究员是论文的第一和通讯作者,复旦大学化学系博士生马睿韬对本工作做出重要贡献。该工作还得到了中科院大连化学物理研究所张东辉院士和美国埃默里大学Joel Bowman教授的关键反馈,并得到国家自然科学基金委的项目支持。
全文链接:https://www.nature.com/articles/s43588-025-00790-0


